Haematology expert Professor Barbara Bain looks at morphology and the accurate diagnosis of the myelodysplastic syndromes.
The myelodysplastic syndromes (MDS) are a group of clonal, neoplastic conditions characterised by ineffective and dysplastic haemopoiesis and a tendency to evolve into acute myeloid leukaemia. Ineffective haemopoiesis is recognised from the usual discrepancy between a hypercellular bone marrow and peripheral cytopenia. It is the result of an increased rate of death of haemopoietic cells by apoptosis within the bone
marrow. Dysplasia describes cytologically abnormal cells in the peripheral blood, bone marrow or both. There are other causes of ineffective haemopoiesis (e.g. megaloblastic anaemia) and other causes of dysplastic haemopoiesis (e.g. exposure to toxic substances and administration of certain drugs and growth factors, HIV infection, copper deficiency and certain inherited conditions).
Accurate diagnosis of MDS can require not only a clinical history, examination of the patient and assessment of the peripheral blood count and film but also examination of the bone marrow morphology and cytogenetic/molecular analysis. The role of peripheral blood and bone marrow morphology is crucial but an assessment of the patient is also important, to distinguish MDS from some of the conditions that can resemble it.
Peripheral blood
The blood count shows cytopenia (one or more of anaemia, neutropenia and thrombocytopenia) and in addition can show an increased MCV and a bimodal histogram of red cell size (in cases with sideroblastic erythropoiesis).
In the blood film, hypogranular or pseudo-Pelger-Huët neutrophils are valuable signs of dysplasia (but also occur in inherited specific granule deficiency and the Pelger-Huët anomaly, respectively). Blast cells can be present in small numbers. Less common dysplastic forms are macropolycytes and granulocytes with pseudo-Chédiak‒Higashi granules. Macrocytosis is less specific than neutrophil anomalies since it is common in liver disease and with excess alcohol intake. However the presence of both normochromic macrocytes and hypochromic microcytes is strongly suggestive of MDS with sideroblastic erythropoiesis. In such cases a search should be made for Pappenheimer bodies. Platelet abnormalities that can be seen include large and giant platelets and hypogranular platelets; both can also occur in inherited conditions but usually with isolated thrombocytopenia rather than with coexisting anaemia or neutropenia.
Bone marrow morphology
In the majority of cases, the bone marrow is hypercellular. Possible dyserythropoietic features include nuclear irregularity or lobulation, bi- or multinuclearity and megaloblastosis (nuclear immaturity in relation to the degree of maturation of the cytoplasm). Erythropoiesis can also be macronormoblastic – erythroblasts are larger than normal but without asynchrony between nuclear and cytoplasmic maturation. In patients with sideroblastic erythropoiesis, vacuolation and defective haemoglobinisation of erythroblasts may be apparent; an iron stain showing ring sideroblasts is confirmatory (but remembering that sideroblastic erythropoiesis can also be due to lead poisoning, copper deficiency and inherited sideroblastic anaemia). The neutrophil series can show hypogranularity of precursors, due to reduced primary granules, as well as of mature cells. There can be a “left shift” in both erythropoiesis and granulopoiesis with immature cells being present as a disproportionately high percentage; this is the morphological correlate of ineffective haemopoiesis. The presence of micromegakaryocytes provides very specific evidence of a haematological neoplasm so is useful in the diagnosis of MDS. These cells are about the size of a promyelocyte with a small nucleus and platelet-type granules in the cytoplasm. Less specific, but also diagnostically useful, is the presence of megakaryocytes that are mainly of normal size but are hypolobated. Increased numbers of such cells suggests the possibility of deletion of part of the long arm of chromosome 5, this type of MDS being popularly known as the “5q‒ syndrome”.
“ The presence of micromegakaryocytes provides very specific evidence of a haematological neoplasm”
Differential diagnosis
The differential diagnosis of MDS includes megaloblastic anaemia due to deficiency of vitamin B12 or folic acid. Both MDS and megaloblastic anaemias are characterised by ineffective and dysplastic haemopoiesis with a hypercellular bone marrow and peripheral cytopenia. The blood film is useful in making the distinction since oval macrocytes and hypersegmented neutrophils are much more common in the vitamin deficiencies than in MDS. Bone marrow morphology is also useful; although megaloblastic erythropoiesis can be a feature of MDS, giant metamyelocytes are quite uncommon as a feature. Misdiagnosis of megaloblastic anaemia as MDS is a very serious error, which usually results from not considering the possibility of a vitamin deficiency.
Other causes of dyserythropoiesis include congenital dyserythropoietic anaemia, sickle cell disease (particularly in conditions of “haemopoietic stress”), thalassaemia, various infections (including malaria and leishmaniasis) and arsenic toxicity. Ring sideroblasts can result from lead poisoning and from administration of various drugs, including isoniazid.
Other causes of dysgranulopoiesis include the inherited conditions mentioned above and also myelokathexis (an inherited condition with morphologically abnormal granulocytes). Acquired conditions of relevance include HIV infection, administration of G-CSF (granulocyte colony-stimulating factor) and the effects of various drugs including chemotherapeutic and immunosuppressive agents. Drugs that can cause a Pelger-Huët anomaly include tacrolimus and mycophenolate mofetil; the presence of detached nuclear fragments, resembling the Howell-Jolly bodies of red cells, can be a clue to this diagnosis.
Copper deficiency (including that due to zinc excess) can simulate MDS by causing pancytopenia, macrocytosis, neutropenia and sideroblastic erythropoiesis; anaemia can also be microcytic or normocytic.
A clue to the correct diagnosis is the presence of vacuolation of haemopoietic precursors on bone marrow examination. Chemotherapeutic agents can simulate MDS by causing pancytopenia and dysplasia; in the case of folic acid antagonists and drugs interfering more directly with DNA synthesis, there is megaloblastosis. Usually the cause is evident from the history but with patients transferring between hospital or even countries, the clinical history may be inadequate.
Conclusion
The morphologist can play a crucial role in the diagnosis of MDS. Often the clinical history provided to the laboratory is not really adequate and a telephone call or reference to the patient’s electronic records may be necessary to clarify the situation. It is important not only to raise the suspicion of MDS but to be constantly aware of the possibility of conditions that can simulate it. Patients with leishmaniasis and vitamin B12 deficiency have been misdiagnosed and treated as MDS, the latter even occasionally being referred for transplantation.
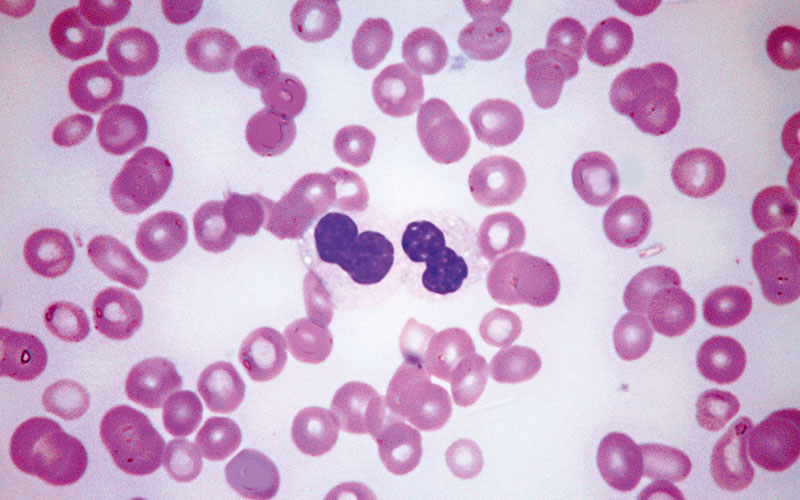
Fig. 1. Peripheral blood film showing two hypogranular neutrophils, which also have nuclei of abnormal shapes. Note also, severe thrombocytopenia. May–Grünwald–Giemsa, high power.
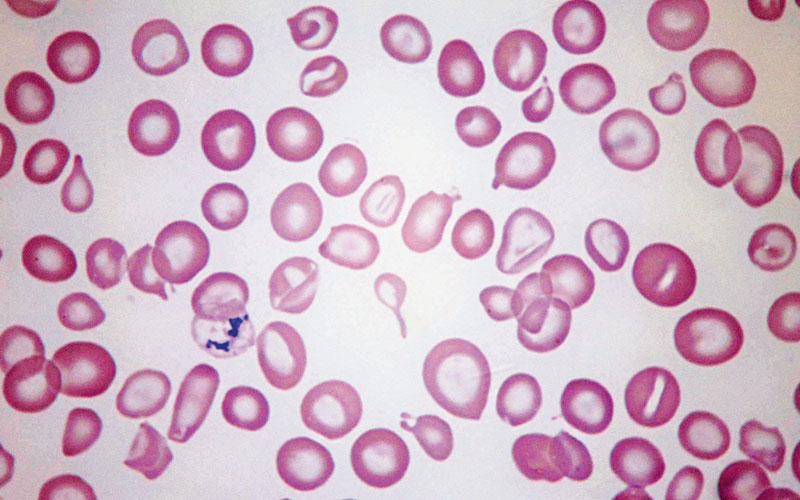
Fig. 2. Peripheral blood film in myelodysplastic syndrome with sideroblastic erythropoiesis showing anisocytosis (macrocytes and microcytes with some hypochromic microcytes but not clearly dimorphic), poikilocytosis (including teardrop poikilocytes and stomatocytes) and an erythrocyte containing unusually large Pappenheimer bodies. May–Grünwald–Giemsa, high power.
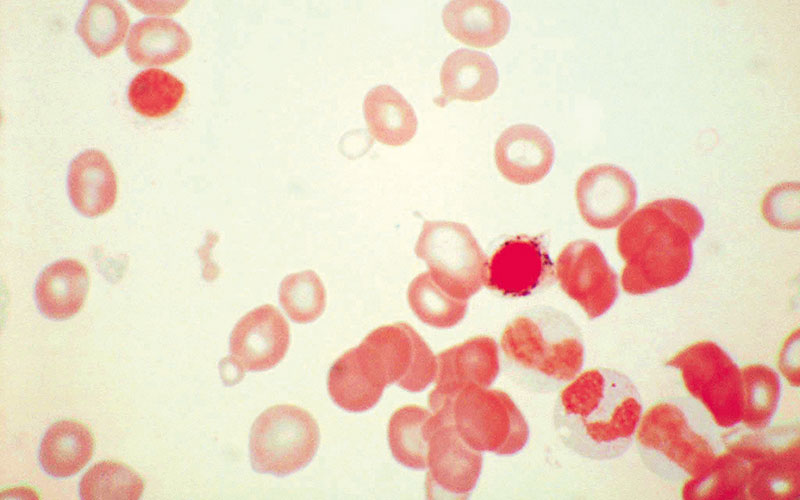
Fig. 3. Bone marrow film in myelodysplastic syndrome with sideroblastic erythropoiesis showing a ring sideroblast. Prussian blue stain, high power.
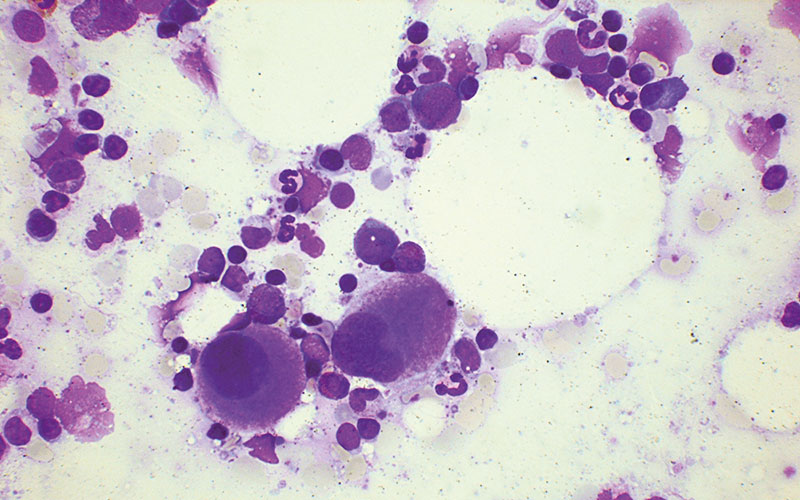
Fig. 4. Bone marrow film in myelodysplastic syndrome with isolated 5q− showing two megakaryocytes with non-lobed nuclei. May–Grünwald–Giemsa, low power.
Barbara Bain is Professor of Diagnostic Haematology at Imperial College London and a consultant at St Mary’s Hospital, London. Professor Bain is the interviewee on this months IBMS podcast, where she discusses a range of topics, from her route into the profession and her work in diagnostic haematology, to her love of Italian art and her personal collection of images of the martyr St Sebastian.



