Clinical Scientist Matthew Hopkins gives a primer on fetal/neonatal alloimmune thrombocytopenia and the screening and treatment options available.
Fetal/neonatal alloimmune thrombocytopenia (FNAIT) is a rare but potentially life-threatening condition that can cause severe thrombocytopenia in the fetus in utero, or neonate postpartum. Intracranial haemorrhage (ICH), which can occur in 10-20% of cases, is the most damaging complication of FNAIT and can cause severe disability or even death.
Incidence in a Caucasian population is estimated to be 1:1000-1:5000 live births, although it appears to be under-diagnosed. The condition manifests due to fetomaternal incompatibility against human platelet antigens (HPA). That is, platelet-specific antigens inherited from the father that are absent in the mother cause an immune response generating maternally-derived antibodies (IgG) that can lead to destruction of fetal platelets. FNAIT can be considered the platelet equivalent of haemolytic disease of the fetus/newborn (HDFN) but, unlike HDFN, FNAIT can occur primigravidae due to transplacental passage of fetal platelets early in pregnancy. Maternal HPA alloantibodies are the most common cause of severe thrombocytopenia (<50x109/L) (normal range: 150-450x109/L) in an otherwise healthy neonate.
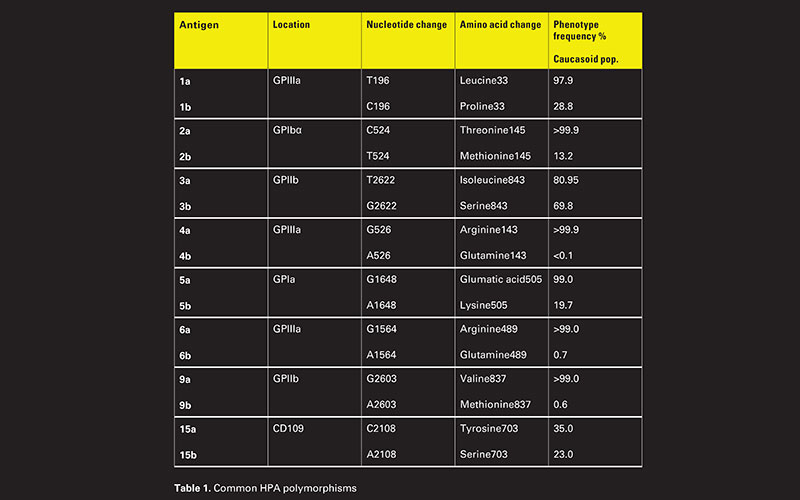
Human platelet antigens are localised to either platelet glycoproteins (GP)IIb/IIIa, GPIa/IIa, GPIb/IX/V or to GPI-linked anchor membrane molecule CD109. Most alloantigens are bi-allelic and result from single nucleotide polymorphisms (SNP) (see Table 1 below).
A diagnosis of FNAIT is confirmed serologically by detection of maternally derived alloantibodies in an alloantigen-negative mother, where the child has inherited the cognate alloantigen from the father. Antibodies directed to HPA-1a and HPA-5b are responsible for approximately 95% of serologically confirmed cases of FNAIT in a Caucasian population; alloantibodies to HPA-1a are implicated in approximately 80% of these cases and 15% are due to HPA-5b alloantibodies.
Currently, 29 different HPA polymorphisms have been described; however, alloantibodies to HPAs that are not HPA-1 to -5 or HPA-15 are rare, often appearing within a single family (low frequency antigens). The rarity of these alloantigens means that testing is not performed routinely, but can be identified by cross-matching paternal platelets with maternal serum if there is a strong clinical suspicion of FNAIT. Laboratory testing
The Histocompatibility and Immunogenetics (H&I) laboratory at NHSBT Filton is an international reference centre for platelet and granulocyte immunology testing. The laboratory provides specialist tests for the detection and identification of platelet-specific antibodies. The standard testing protocol in our laboratory comprises two “in-house” assays: the platelet immunofluorescence test (PIFT) and monoclonal immobilisation of platelet antigens (MAIPA), using suitably HPA typed platelet donor cell panels (HPA-1, 2, 3, 5 and 15).
The PIFT uses a flow cytometric end point to detect bound antibodies (IgG) to the platelet membrane. This test can detect both HPA-specific and non-specific antibodies. The MAIPA is a sandwich enzyme-linked immunosorbent assay (ELISA) that uses glycoprotein-specific mouse monoclonal antibodies to capture fragments of platelet membrane expressing HPA epitopes.
Bound human IgG is visualised using a horseradish peroxidase-conjugated immunoglobulin that reacts with its substrate, promoting a colour change within the microplate. This colour change is analysed in order to visualise HPA-specific antibodies. Testing can be further augmented, if required, by the use of a bead-based assay (Immucor, Norcross, GA) which utilises Luminex x-map technology. The assay uses polystyrene beads coated with HPA antigens as a target. Anti-human IgG conjugated with a fluorochrome is used to identify if platelet-specific antibodies are present.
Additionally, the parents and infant (if available) are HPA genotyped by amplifying DNA using polymerase chain reaction-sequence based typing (PCR-SBT) with “in-house” HPA-1-6, -9, and -15 primers. Identifying the zygosity of the father can aid in predicting FNAIT risk (see Figure 1 below).
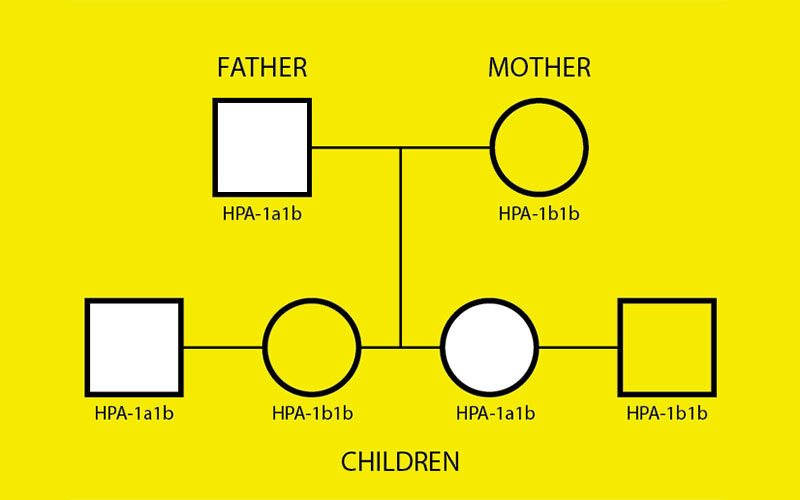
Figure 1. In this example, the mother is HPA-1b1b and the father is HPA-1a1b. The children may be either HPA-1a1b or HPA-1b1b depending on whether the baby inherits HPA-1a or HPA-1b from the father. There is a 50% chance that a child will inherit HPA-1a and be at risk of FNAIT.
Treatment options
Since FNAIT can occur in first pregnancies, it is often identified post-delivery, usually presenting with a low platelet count with or without external petechiae/purpura, or bleeding. A platelet count should be performed and if severe isolated thrombocytopenia is evident (<50x109/L) in an otherwise healthy neonate, then FNAIT should be suspected and investigated. Platelet support for the neonate should not be delayed while waiting for confirmation from laboratory tests.
The 2013 Guidelines for the Blood Transfusion Services in the UK advises that neonatal platelet counts should be maintained above 30x109/L or 100x109/L if intracranial haemorrhage (ICH) is evident. NHSBT provides specialist stock of “on-the-shelf” neonatal platelet components which are phenotypically HPA-1a(-) -5b(-). These products are held in strategically placed holding centres throughout England and should be effective in 95% of FNAIT cases due to the prevalence of HPA-1a and HPA-5b antibodies. In subsequent pregnancies for women who are alloimmunised, FNAIT can often be more severe. These pregnancies are closely monitored by regular ultrasounds for any signs of fetal abnormalities, such as ICH or ventriculomegaly that might indicate thrombocytopenia. The mother will also be screened regularly throughout her pregnancy. Fetal HPA genotyping from amniotic fluid can also be performed to confirm whether the antigen has been inherited from the father and thus calculate the risk of FNAIT. First-line antenatal treatment for mothers who are alloimmunised is intravenous immunoglobulin G (IvIgG) (1g/kg body weight at weekly intervals) usually used in isolation or in conjunction with corticosteroids.
Intrauterine transfusion (IUT) of HPA-1a(-) -5b(-) platelet hyperconcentrates can be given while fetal blood sampling is performed. These products only have a shelf life of 24 hours and the laboratory requires at least one week’s notice in order to arrange a suitable blood donor to provide this product. Due to a high risk of fetal morbidity and mortality, this treatment option should only be considered if the first-line treatment therapy fails.
Matthew Hopkins is a Clinical Scientist working in Histocompatibility and Immunogenetics at NHS Blood and Transplant, Filton.
To see the article with full references, visit thebiomedicalscientist.net